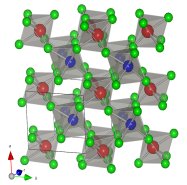
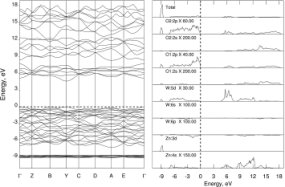
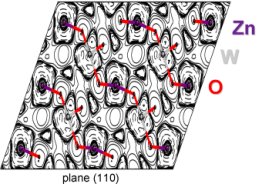
Quantum Chemistry (QC) is a branch of science which uses quantum mechanics to
treat chemical and physical properties of compounds on atomic level.
In our laboratory, we use it to predict the structure of polycrystalline and nanocrystalline
materials and to interpret Raman and XANES spectra.
USEFUL LINKS
Wikipedia: Quantum chemistry.
PRACE - partnership for advanced computing in Europe.
Crystal - a computational tool for solid state chemistry and physics.
CRYSPLOT - an online web-oriented tool to visualize computed properties of periodic systems.
Basis Set Exchange from EMSL.
Pseudopotentials, ECPs at Uni-Stuttgart.
VASP - a package for performing ab-initio quantum-mechanical molecular dynamics (MD) using pseudopotentials and a plane wave basis set.
ABINIT - Density Functional Theory (DFT) using pseudopotentials and a planewave or wavelet basis.
Quantum-Espresso - a code based on density-functional theory, plane waves, and pseudopotentials for electronic-structure calculations and materials modeling at the nanoscale.
BURAI - a GUI for Quantum-Espresso.
QuantumVITAS - a Quantum Visual Interactive Toolkit for Ab-initio Simulations based on Quantum-Espresso.
Exciting - a full-potential all-electron density-functional-theory package implementing the families of linearized augmented planewave methods.
CASTEP - a first-principles code based on plane-wave basis set and pseudopotentials.
Wien2k - a first-principles code based on full-potential (linearized) augmented plane-wave ((L)APW) + local orbitals (lo) method.
SIESTA - a code to perform efficient electronic structure calculations and ab initio molecular dynamics simulations of molecules and solids.
LmtART - the full-potential linear-muffin-tin-orbital (FP-LMTO) program to perform band structure, total energy and force calculations within the methods of density functional theory (DFT).
Elk - an all-electron full-potential linearised augmented-plane wave (FP-LAPW) code.
ORCA - an ab initio, DFT and semiempirical SCF-MO package.
ASE - is a set of tools and Python modules for setting up, manipulating, running, visualizing and analyzing atomistic simulations.
Quantum Mobile - a Virtual Machine for computational materials science.
Bilbao Crystallographic Server.
ISOTROPY - a software package which applies group theoretical methods to the analysis of phase transitions in crystalline solids.
FINDSYM - identifies the space group of a crystal, given the positions of the atoms in a unit cell.
USPEX - universal structure predictor.
CALYPSO - an efficient code for structure prediction.
CrGraph - visualization of computed properties (bands, DOS, charge density, electrostatic potential) for Crystal.
MolDraw - a program to display and manipulate molecular and crystalline structures.
XCrySDen - A crystalline and molecular structure visualisation program.
J-ice - a Jmol Interface for Crystallographic and Electronic Properties.
Gabedit - a graphical user interface to computational chemistry packages like Gamess-US, Gaussian, Molcas, Molpro, MPQC, OpenMopac, Orca, PCGamess and Q-Chem.
Ascalaph - a molecular modelling suite.
VESTA - a 3D visualization program for structural models and 3D grid data such as electron/nuclear densities.
Mercury - a comprehensive range of tools for 3D structure visualisation and the exploration of crystal packing.
VMD - a molecular visualization program for displaying, animating, and analyzing large biomolecular systems using 3-D graphics and built-in scripting.
POV-Ray - the Persistence of Vision Raytracer, a tool for producing high-quality computer graphics.
DL-FIND - a geometry optimiser for quantum chemical and QM/MM calculations.
Phonopy - an open source phonon analyzer.
ASE - Atomic Simulation Environment.
Quantum Wise - simulation software for nanoscience.
Tilde - distributed database of physico-chemical properties calculated ab initio (use Firefox !).
ToposPro - a program package for comprehensive analysis of geometrical and topological properties of periodic structures (crystals, networks, tilings).
MaterialsCloud - useful tools for computational materials science.
ETSF - European Theoretical Spectroscopy Facility.
Materials Project - searchable materials database at Lawrence Berkeley National Laboratory.
NOMAD Repository.
PSI-K - a Europe-based, worldwide network of researchers working on the advancement of first-principles computational materials science.
|
 Large Visitor Map
Large Visitor Map