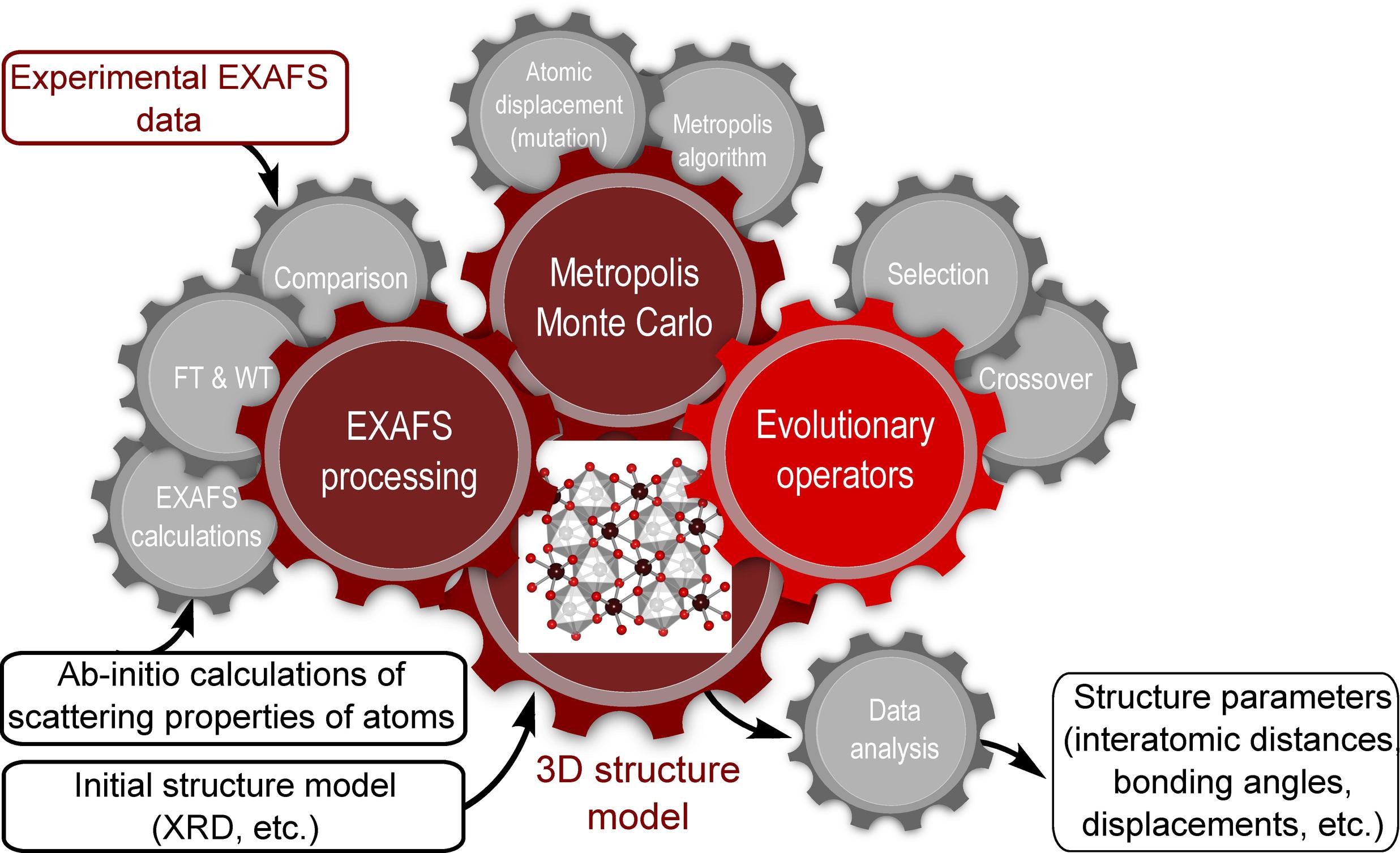
EvAX (Evolutionary Algorithms for XAS analysis) is a program for the analysis of extended X-ray absorption fine structure (EXAFS) data [1] using reverse Monte Carlo (RMC) and evolutionary algorithm (EA) methods. It is designed to be especially useful for studies of materials with well-defined structure, such as crystalline and nanocrystalline materials. Some of the main strengths of EvAX method are:
Analysis of multiple scattering effects: to benefit from structural information hidden in the contributions of multiple-scattering (MS) effects and of distant coordination shells, EvAX code provides great flexibility for treating all important scattering paths with user-specified precision.
Evolutionary algorithm-based structure optimization: the optimization process that employs the power of evolutionary algorithm allows much more efficient exploration of the possible configuration space and makes feasible advanced analysis of complex compounds with low symmetry.
Usage of wavelet transform for EXAFS signal comparison: the representation of EXAFS spectra in k- and R-spaces simultaneously using continuous Morlet wavelet transform allows one to obtain more information from the same experimental data and to have better control over the difference between the experimental and calculated EXAFS data.
Analysis of EXAFS data from several absorption edges simultaneously: EvAX code allows one to construct an unambiguous single structure model, consistent with EXAFS data acquired at several absorption edges.
EvAX can be used both with Windows (Windows 7 and newer) and Linux operating systems. The most time-consuming parts of calculations can be automatically divided into smaller problems and performed in parallel (in different threads of execution), taking advantage of multi-processor systems.
EvAX code is designed for the RMC-like analysis of well-defined structures. Unlike it is in some other implementations of RMC-method, EvAX allows only small atomic displacements of atoms from their initial positions. Hence, it enables the modelling of complex bond-length distributions and allows one to analyze changes in interatomic distances and structural disorder, but does not allow one to refine the coordination numbers. The latter are imposed by the initial structure model provided by user, and are not changed during the EvAX simulations. Therefore EvAX is useful for the cases, where coordination numbers are known in advance, e.g., for the analysis of disorder in crystalline materials or well-defined nanocrystalline materials.
Here are two possible scenarios, when EvAX can be useful:
(i) You know the approximate structure of the material (e.g., from X-ray diffraction), and you want to use EXAFS data to study the disorder effects and atomic thermal motion.
(ii) You have several plausible structure models for your material (e.g., from DFT simulations), and you want to know, which of them fits better the available experimental EXAFS data.
Besides its main purpose - RMC-like analysis - EvAX can also be used as an efficient tool for calculations of theoretical EXAFS spectra for a given low-dimensional atomic configuration or a set of such configurations, generated, e.g., by some molecular dynamics routine. It can also be used for efficient calculations of wavelet transforms, Fourier transform and inverse Fourier transforms for given EXAFS spectra and generation of input files for FEFF code.
EvAX is a command-line program. For calculations following files and programs should be prepared:
EvAX program, compiled for a given operating system.
FEFF program (version 8 or higher), compiled for a given operating system (FEFF code [1] can be ordered and downloaded from FEFF project webpage. FEFF version 8.5L is available free of charge and is sufficient for EvAX calculations).
For calculations under Linux OS - the auxiliary runFEFF.exe script file is needed (see the manual).
Parameter file - text file that contains the values of the parameters for calculations
Structure file - text file that defines the lattice and types of atoms, and the initial positions of atoms in the structure. Software like VESTA program can be very helpful in preparation of this file.
File(s) with experimental extracted EXAFS data with subtracted background. EvAX can work directly with EXAFS data, preprocessed by such codes as ATHENA or EDA.
The names and formats of structure file, file with experimental EXAFS data and path to FEFF program are specified within the parameter file. The name of parameter file is supplied to EvAX program as a command-line argument. Thus, to start calculations, just type
EvAX.exe parameters.dat
For Linux OS, of course, slightly different commands needs to used:
./EvAX.exe parameters.dat
An example of parameter file can be generated using following command:
EvAX.exe -example
This will generate a file example.dat with the default values for all calculation parameters. -example is not the only one additional command that can be supplied to EvAX via command line. Other commands are discussed below. A brief description of these commands (flags) can also be obtained by running
EvAX.exe -help